s |
focus search bar ( enter to select, ▲ / ▼ to change selection) |
g c |
go to cluster |
g e |
go to edctools |
g f |
go to facility |
g g |
go to guidelines |
h |
toggle this help ( esc also exits) |
iPOP-UP training: hands-on
Date: 12/06/2023
Trainers: Olivier Kirsh, Julien Rey, Magali Hennion
Presentation
The slides of the presentation can be downloaded here.
Table of content
- Connect to the cluster
- Optional: use a file explorer
- Get information about the cluster
- Slurm sbatch command
- Useful sbatch options 1/2
- Modules
- Job handling and monitoring
- Practical example
- Parallelization
- Useful sbatch options 2/2
- Multi-threading
- Use Slurm variables
- Job arrays
- Job arrays examples
- Job Array Common Mistakes
- Complex workflows
- Useful resources
- Thanks
Connect to the cluster
ssh -o PubkeyAuthentication=no username@ipop-up.rpbs.univ-paris-diderot.fr
Warm-up
Where are you on the cluster?
pwd
Then explore the /shared folder
tree -L 1 /shared
/shared/banks folder contains commonly used data and resources. Explore it by yourself with commmands like ls or cd.
Can you see the first 10 lines of the mm10.fa file? (mm10.fa = mouse genomic sequence version 10)
There is a training project accessible to you, navigate to this folder and list what is inside.
Then go to one of your projects and create a folder named 230612_training. This is where you will do all the exercices.
Optional: use a file explorer
Using the file manager from GNOME, you can navigate easily on iPOP-UP file server.
- Open the file manager
Fichiers. - Click on
Autres emplacementson the side bar. - In the bar
Connexion à un serveur, typesftp://ipop-up.rpbs.univ-paris-diderot.fr/and press the enter key. - Enter your login and password.
This way, you can modify your files directly using any local text editor.
Get information about the cluster
sinfo
Slurm sbatch command
sbatch allows you to send an executable file to be ran on a computation node.
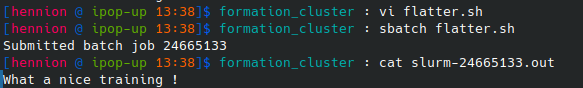
Exercise 1: my first sbatch script
Starting from 01_02_flatter.sh, make a script named flatter.sh printing “What a nice training !”
Then run the script:
sbatch flatter.sh
The output that should have appeared on your screen has been diverted to slurm-xxxxx.out but this name can be changed using SBATCH options.
Exercise 2: my first SBATCH option
Modify flatter.sh to add this line:
#SBATCH -o flatter.out
then run it. Anything different ?
Exercise 3: hostname
Run using sbatch the command hostname in a way that the sbatch outfile is called hostname.out.
What is the output ? How does it differ from typing directly hostname in the terminal and why ?
Useful sbatch options 1/2
| Options | Flag | Function |
|---|---|---|
| −−partition | -p | partition to run the job (mandatory) |
| −−job-name | -J | give a job a name |
| −−output | -o | output file name |
| −−error | -e | error file name |
| −−chdir | -D | set the working directory before running |
| −−time | -t | limit the total run time (default : no limit) |
| −−mem | memory that your job will have access to (per node) |
To find out more, the Slurm manual man sbatch or https://slurm.schedmd.com/sbatch.html.
Modules
A lot of tools are installed on the cluster. To list them, use one of the following commands.
module available
module avail
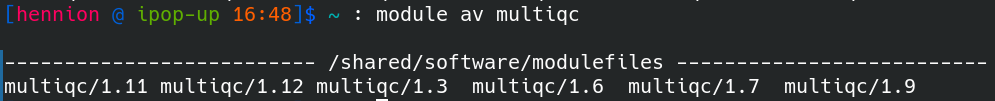
module av
You can limit the search for a specific tool, for example look for the different versions of multiqc on the cluster using module av multiqc.
To load a tool
module load tool/1.3
module load tool1 tool2 tool3
To list the modules loaded
module list
To remove all loaded modules
module purge
Job handling and monitoring
Exercise 4: follow your jobs
The sleep command : do nothing (delay) for the set number of seconds.
Restart from 03_04_hostname_sleep.sh and launch a simple job that will launch sleep 600.
squeue
On your terminal, type
squeue
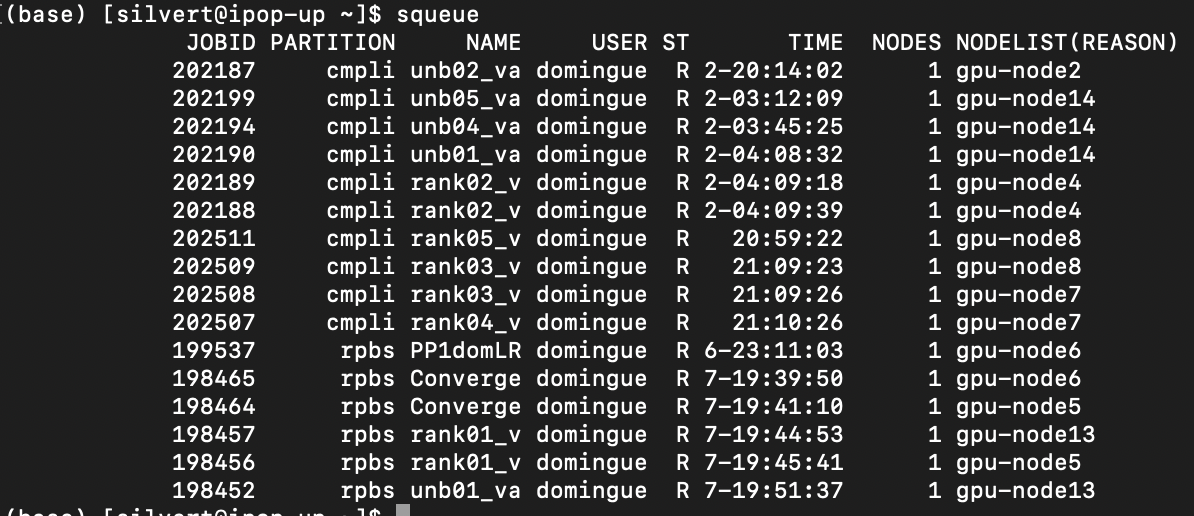
ST Status of the job.
R = Running
PD = Pending
To see only iPOP-UP jobs
squeue -p ipop-up
To see only the jobs of untel
squeue -u untel
To see only your jobs
squeue --me
scancel
To cancel a job which you started, use the scancel command followed by the jobID (Number given by SLURM, visible in squeue)
scancel jobID
You can stop the previous sleep job with this command.
sacct
Re-run sleep.sh and type
sacct
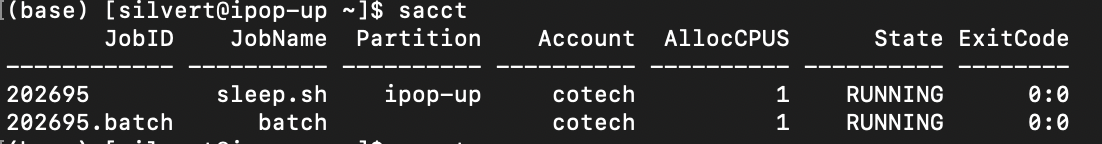
You can pass the option --format to list the information that you want to display, including memory usage, time of running,…
For instance
sacct --format=JobID,JobName,Start,Elapsed,CPUTime,NCPUS,NodeList,MaxRSS,ReqMeM,State
To see every options, run sacct --helpformat
Job efficiency : seff
After the run, the seff command allows you to access information about the efficiency of a job.
seff <jobid>
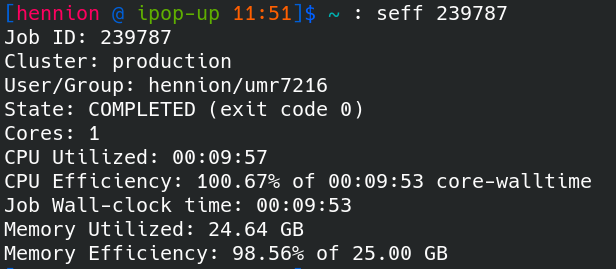
Practical example
Exercise 5 : Alignment
Run an alignment using STAR version 2.7.5a starting from 05_06_star.sh.
- The FASTQ files to align are in
/shared/projects/training/test_fastq. - You need an index folder for STAR (version 2.7.5a) for the mouse mm39 genome, look for it in the banks.
- You have to increase the RAM to 25G.
After the run
Check the resource that was used using seff.
Parallelization
Useful sbatch options 2/2
| Options | Default | Function |
|---|---|---|
| −−nodes | 1 | Number of nodes required (or min-max) |
| −−nodelist | Select one or several nodes | |
| −−ntasks-per-node | 1 | Number of tasks invoked on each node |
| −−mem | 2GB | Memory required per node |
| −−cpus-per-task | 1 | Number of CPUs allocated to each task |
| −−mem-per-cpu | 2GB | Memory required per allocated CPU |
| −−array | Submit multiple jobs to be executed with identical parameters |
Multi-threading
Some tools allow multi-threading, i.e. the use of several CPUs to accelerate one task. It is the case of STAR with the --runThreadN option.
Exercise 6: Alignment, parallel
Modify the previous sbatch file to use 4 threads to align the FASTQ files on the reference. Run and check time and memory usage.
Use Slurm variables
The Slurm controller will set some variables in the environment of the batch script. They can be very useful. For instance, you can improve the previous script using $SLURM_CPUS_PER_TASK.
The full list of variables is visible here.
Some useful ones:
- $SLURM_CPUS_PER_TASK
- $SLURM_JOB_ID
- $SLURM_JOB_ACCOUNT
- $SLURM_JOB_NAME
- $SLURM_JOB_PARTITION
Of note, Bash shell variables can also be used in the sbatch script:
- $USER
- $HOME
- $HOSTNAME
- $PWD
- $PATH
Job arrays
Job arrays allow to start the same job a lot of times (same executable, same resources) on different files for example. If you add the following line to your script, the job will be launch 6 times (at the same time), the variable $SLURM_ARRAY_TASK_ID taking the value 0 to 5.
#SBATCH --array=0-5
Exercice 7 : Job array
Starting from 07_08_array_example.sh, make a simple script launching 6 jobs in parallel.
Exercice 8 : fair resource sharing
It is possible to limit the number of jobs running at the same time using %max_running_jobs in #SBATCH --array option.
Modify your script to run only 2 jobs at the time.
You will see using squeue command that some of the tasks are pending until the others are over.
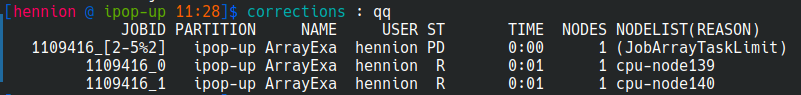
Job arrays examples
Take all files matching a patern in a directory
Example
#SBATCH --array=0-7 # if 8 files to proccess
FQ=(*fastq.gz) #Create a bash array
echo ${FQ[@]} #Echos array contents
INPUT=$(basename -s .fastq.gz "${FQ[$SLURM_ARRAY_TASK_ID]}") #Each elements of the array are indexed (from 0 to n-1) for slurm
echo $INPUT #Echos simplified names of the fastq files
List or find files to process
If for any reason you can’t use bash array, you can alternatively use ls or find to identify the files to process and get the nth with sed (or awk).
#SBATCH --array=1-4 # If 4 files, as sed index start at 1
INPUT=$(ls $PATH2/*.fq.gz | sed -n ${SLURM_ARRAY_TASK_ID}p)
echo $INPUT
Job Array Common Mistakes
- The index of bash arrays starts at 0
- Don’t forget to have different output files for each task of the array
- Same with your log names (\%a or \%J in the name will do the trick)
- Do not overload the cluster! Please use \%50 (for example) at the end of your indexes to limit the number of tasks (here to 50) running at the same time. The 51st will start as soon as one finishes!
- The RAM defined using
#SBATCH --mem=25Gis for each task
Complex workflows
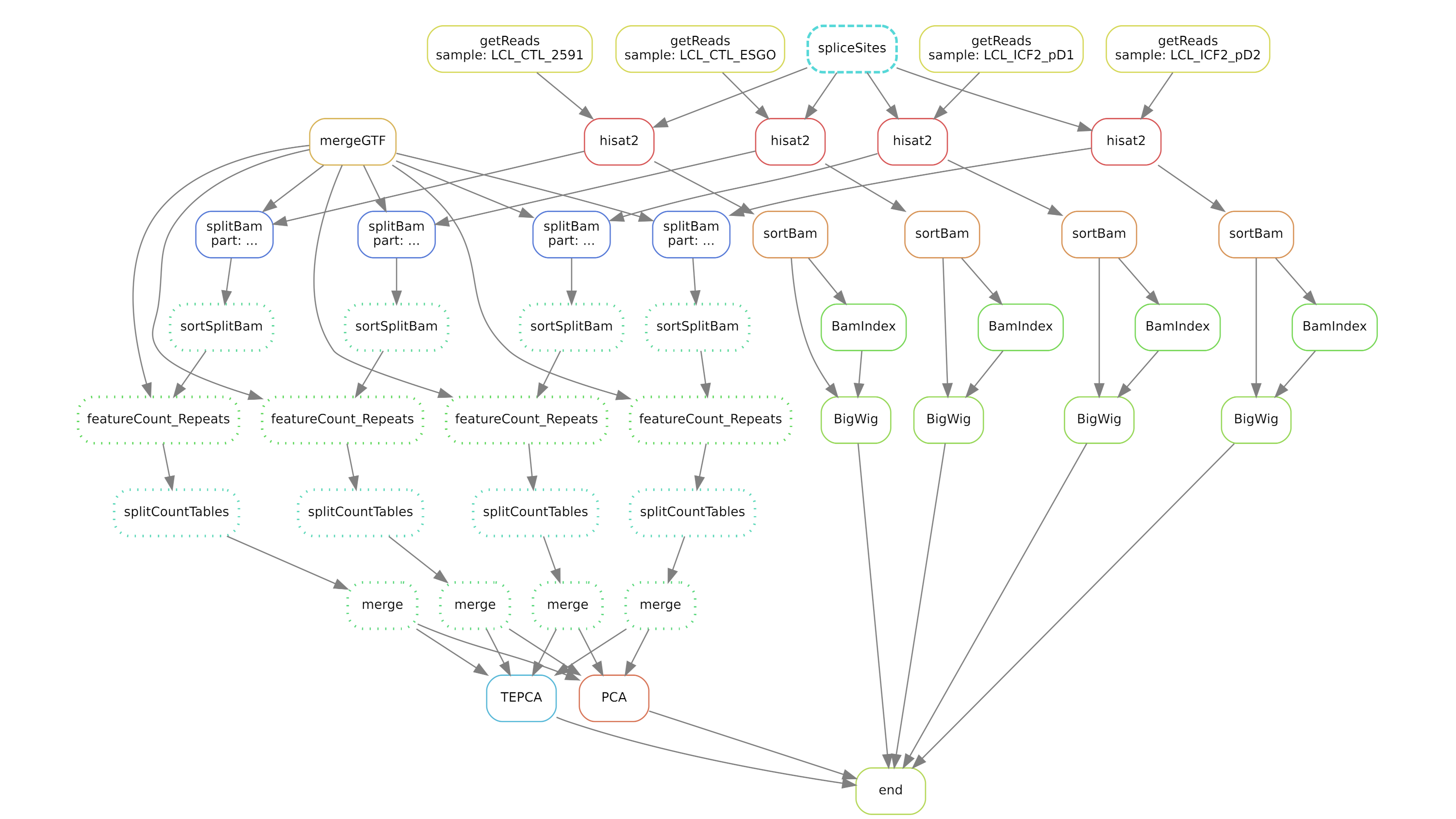
Use workflow managers such as Snakemake or Nextflow.
nf-core workflows can be used directly on the cluster.
Exercice 9: nf-core workflows
Starting from 09_nf-core.sh, write a script running ChIP-seq workflow on nf-core test data.
Some help can be found here. Please also see the full documentation.
Correction
[Of note, even with the test dataset, it takes a lot of time and resources!]
Useful resources
-
To find out more, read the SLURM manual :
man sbatchor https://slurm.schedmd.com/sbatch.html -
Ask for help or signal problems on the cluster : https://discourse.rpbs.univ-paris-diderot.fr/
-
iPOP-UP cluster documentation: https://ipop-up.docs.rpbs.univ-paris-diderot.fr/documentation/
-
BiBs practical guide: https://parisepigenetics.github.io/bibs/cluster/ipopup
-
IFB community support : https://community.france-bioinformatique.fr/
Thanks
- iPOP-UP’s technical and steering committees




|
BiBs
2024 parisepigenetics
https://github.com/parisepigenetics/bibs |
| programming pages theme v0.5.22 (https://github.com/pixeldroid/programming-pages) |